Why Makie.jl?¶
1. Plotting millions of data points is easy¶
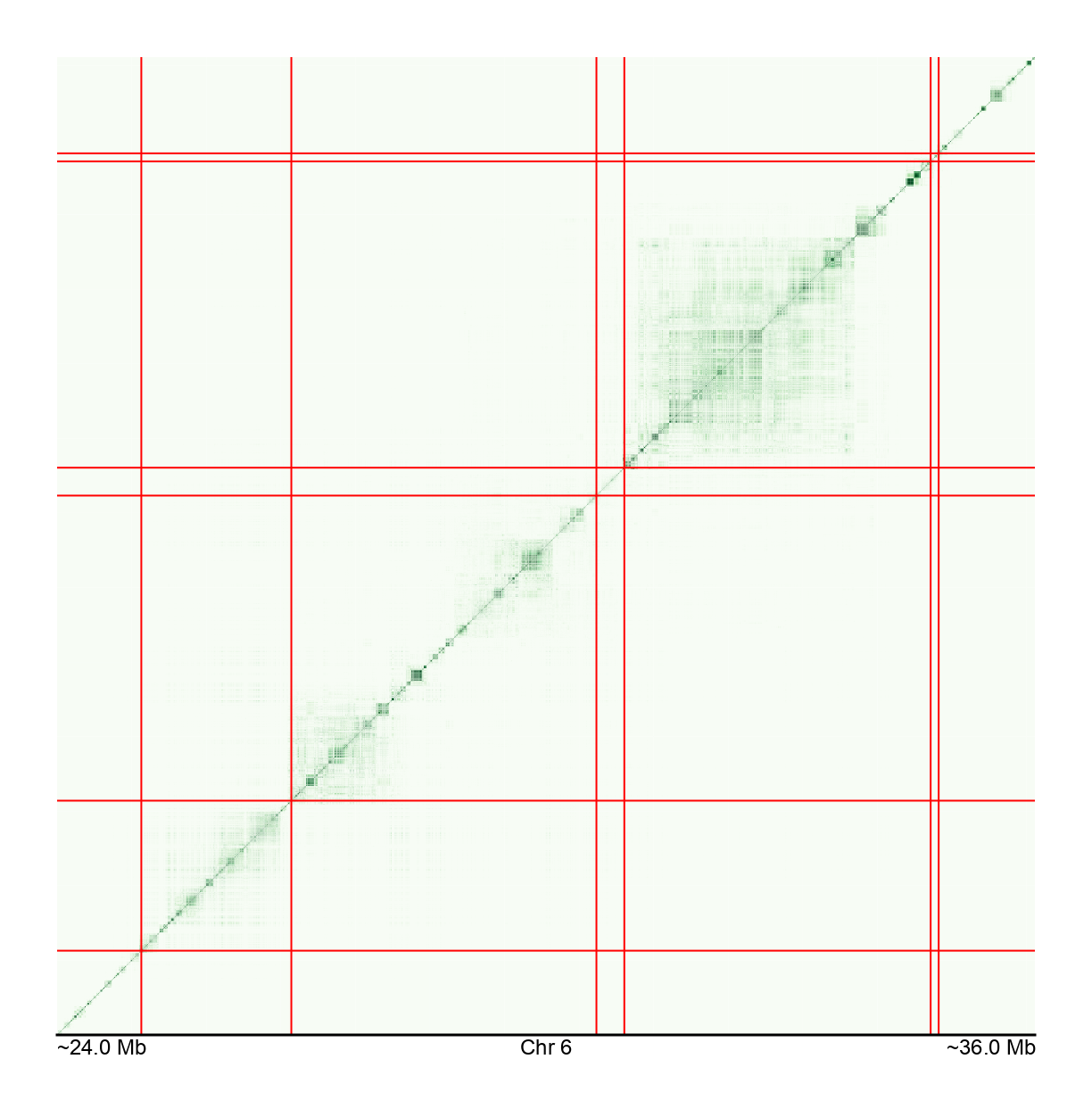
LD structure for ~66,000 SNPs in the MHC region → ≈2 billion unique data points
For more information on the MHC, see Horton et al. 2004
versioninfo()
Julia Version 1.7.0 Commit 3bf9d17731 (2021-11-30 12:12 UTC) Platform Info: OS: macOS (x86_64-apple-darwin19.5.0) CPU: Intel(R) Core(TM) i9-9880H CPU @ 2.30GHz WORD_SIZE: 64 LIBM: libopenlibm LLVM: libLLVM-12.0.1 (ORCJIT, skylake)
LD structure for ~66,000 SNPs in the MHC region → ≈2 billion unique data points
For more information on the MHC, see Horton et al. 2004
Publication-quality figure generated using Makie.jl's default layout tools w/o further modifications (e.g. in Illustrator)

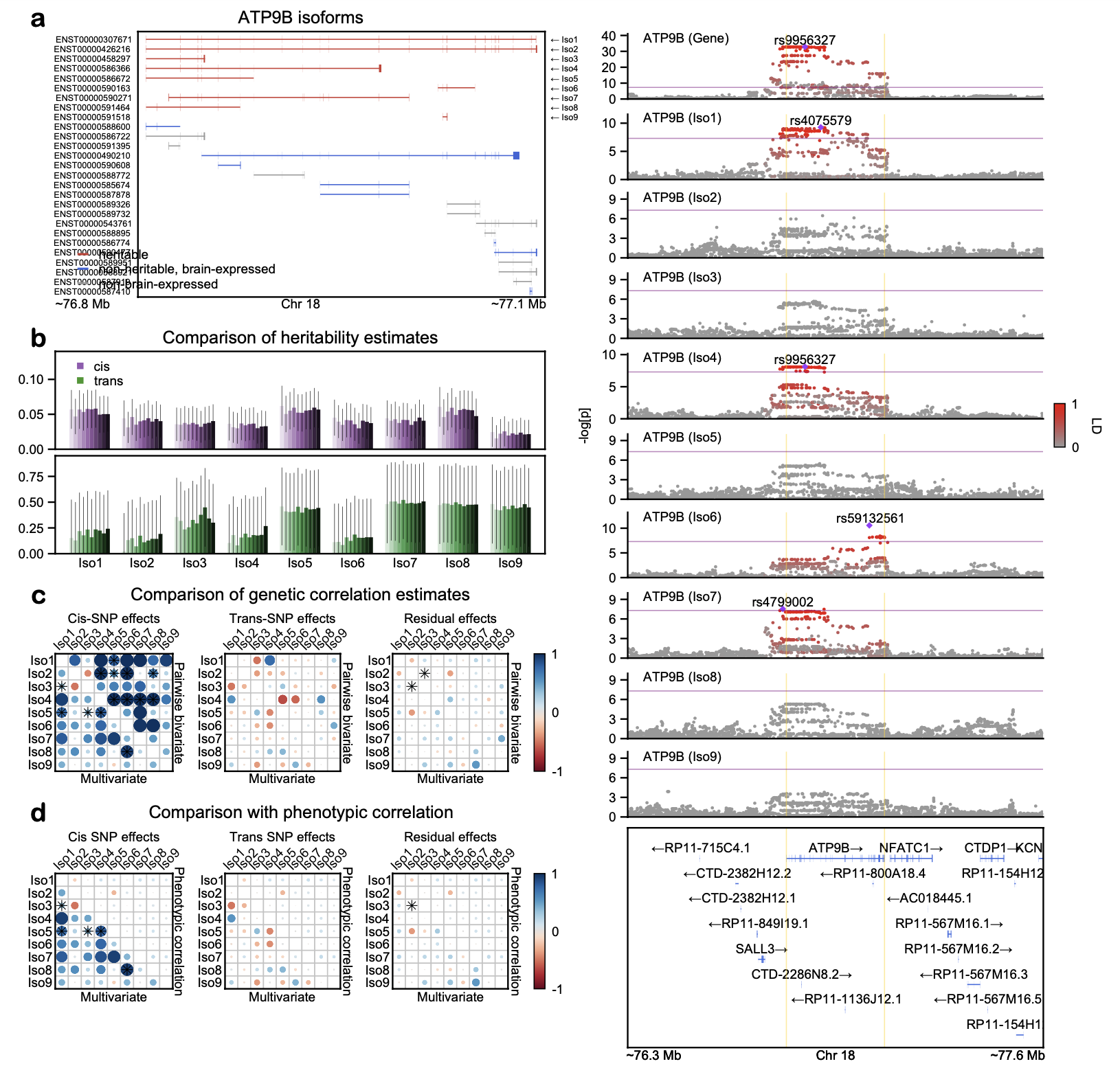
GRIN2A is a high-confidence schizophrenia risk gene
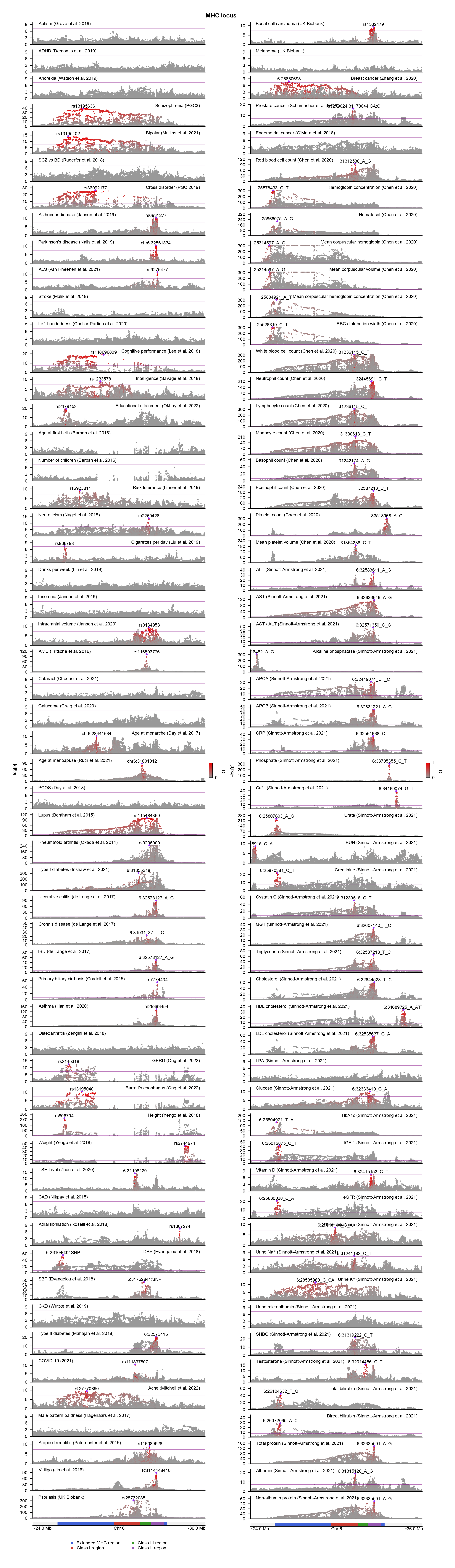
MHC region is one of the most pleiotropic regions in the human genome

Visualizing the backbone of a LocusZoom plot requires genetic association data, gene annotation data, and LD reference panel
using GeneticsMakie, CairoMakie, CSV, DataFrames, SnpArrays
set_theme!(font = "Arial")
@info "Loading GENCODE v39 annotation for chromosome 15"
@time gencode = CSV.read("./gencode.v39lift37.annotation.chr15.gtf.gz", DataFrame,
header = ["seqnames", "source", "feature", "start", "end", "score", "strand", "phase", "info"],
delim = "\t", skipto = 6)
gencode[!, :info]
6.724185 seconds (3.57 M allocations: 269.634 MiB, 1.38% gc time, 94.54% compilation time)
┌ Info: Loading GENCODE v39 annotation for chromosome 15 └ @ Main In[4]:1
117418-element Vector{String}:
"gene_id \"ENSG00000215567.5_1\"; " ⋯ 454 bytes ⋯ "\"; remap_status \"full_contig\";"
"gene_id \"ENSG00000215567.5_1\"; " ⋯ 454 bytes ⋯ "\"; remap_status \"full_contig\";"
"gene_id \"ENSG00000215567.5_1\"; " ⋯ 454 bytes ⋯ "\"; remap_status \"full_contig\";"
"gene_id \"ENSG00000201241.1\"; ge" ⋯ 122 bytes ⋯ "stituted_missing_target \"V38\";"
"gene_id \"ENSG00000201241.1\"; tr" ⋯ 255 bytes ⋯ "stituted_missing_target \"V38\";"
"gene_id \"ENSG00000201241.1\"; tr" ⋯ 299 bytes ⋯ "stituted_missing_target \"V38\";"
"gene_id \"ENSG00000258463.1_1\"; " ⋯ 160 bytes ⋯ "remap_target_status \"overlap\";"
"gene_id \"ENSG00000258463.1_1\"; " ⋯ 404 bytes ⋯ "remap_target_status \"overlap\";"
"gene_id \"ENSG00000258463.1_1\"; " ⋯ 450 bytes ⋯ "\"; remap_status \"full_contig\";"
"gene_id \"ENSG00000274347.1_1\"; " ⋯ 158 bytes ⋯ " 1; remap_target_status \"new\";"
"gene_id \"ENSG00000274347.1_1\"; " ⋯ 404 bytes ⋯ "g\"; remap_target_status \"new\";"
"gene_id \"ENSG00000274347.1_1\"; " ⋯ 454 bytes ⋯ "\"; remap_status \"full_contig\";"
"gene_id \"ENSG00000188403.7_1\"; " ⋯ 166 bytes ⋯ "remap_target_status \"overlap\";"
⋮
"gene_id \"ENSG00000248472.8_1\"; " ⋯ 429 bytes ⋯ "\"; remap_status \"full_contig\";"
"gene_id \"ENSG00000248472.8_1\"; " ⋯ 429 bytes ⋯ "\"; remap_status \"full_contig\";"
"gene_id \"ENSG00000248472.8_1\"; " ⋯ 381 bytes ⋯ "remap_target_status \"overlap\";"
"gene_id \"ENSG00000248472.8_1\"; " ⋯ 429 bytes ⋯ "\"; remap_status \"full_contig\";"
"gene_id \"ENSG00000248472.8_1\"; " ⋯ 429 bytes ⋯ "\"; remap_status \"full_contig\";"
"gene_id \"ENSG00000248472.8_1\"; " ⋯ 429 bytes ⋯ "\"; remap_status \"full_contig\";"
"gene_id \"ENSG00000248472.8_1\"; " ⋯ 461 bytes ⋯ "remap_target_status \"overlap\";"
"gene_id \"ENSG00000248472.8_1\"; " ⋯ 509 bytes ⋯ "\"; remap_status \"full_contig\";"
"gene_id \"ENSG00000248472.8_1\"; " ⋯ 509 bytes ⋯ "\"; remap_status \"full_contig\";"
"gene_id \"ENSG00000248472.8_1\"; " ⋯ 509 bytes ⋯ "\"; remap_status \"full_contig\";"
"gene_id \"ENSG00000248472.8_1\"; " ⋯ 509 bytes ⋯ "\"; remap_status \"full_contig\";"
"gene_id \"ENSG00000248472.8_1\"; " ⋯ 509 bytes ⋯ "\"; remap_status \"full_contig\";"
The :info column in GENCODE gtf file contains rich information about each gene/isoform
GeneticsMakie.parsegtf!(gencode)
names(gencode)
14-element Vector{String}:
"seqnames"
"source"
"feature"
"start"
"end"
"score"
"strand"
"phase"
"info"
"gene_id"
"gene_name"
"gene_type"
"transcript_id"
"transcript_support_level"
select!(gencode, :seqnames, :feature, :start, :end, :strand, :gene_id, :gene_name, :gene_type, :transcript_id)
@assert size(gencode) == (117_418, 9)
@info "Loading 1000 Genomes European reference panel for chromosome 15"
kgp = SnpData("./kgp.chr15")
@assert size(kgp) == (503, 200_311)
┌ Info: Loading 1000 Genomes European reference panel for chromosome 15 └ @ Main In[7]:1
@info "Loading GWAS results for chromosome 15"
gwas = CSV.read("./hernia.chr15.gz", DataFrame, comment = "##", missingstring = "NA")
┌ Info: Loading GWAS results for chromosome 15 └ @ Main In[8]:1
299,624 rows × 8 columns
| CHR | POS | SNPID | Allele1 | Allele2 | AFAllele2 | BETA | p | |
|---|---|---|---|---|---|---|---|---|
| Int64 | Int64 | String | String | String | Float64 | Float64 | Float64 | |
| 1 | 15 | 20001226 | rs28896870 | C | T | 0.113114 | 0.0110027 | 0.487073 |
| 2 | 15 | 20001774 | rs28812614 | T | C | 0.257554 | 0.0216504 | 0.0621705 |
| 3 | 15 | 20004721 | rs145629091 | A | G | 0.137294 | 0.0259993 | 0.0793635 |
| 4 | 15 | 20014120 | rs12594432 | G | A | 0.138131 | 0.0267138 | 0.0689262 |
| 5 | 15 | 20017513 | rs12900040 | T | C | 0.118101 | 0.0100042 | 0.522537 |
| 6 | 15 | 20021591 | rs11535026 | T | A | 0.116481 | 0.00981448 | 0.531855 |
| 7 | 15 | 20021749 | rs12595413 | C | T | 0.137905 | 0.0264183 | 0.0724226 |
| 8 | 15 | 20026191 | rs533345786 | A | AAAG | 0.111671 | 0.0109291 | 0.504701 |
| 9 | 15 | 20026200 | rs543944619 | A | C | 0.112151 | 0.010696 | 0.511987 |
| 10 | 15 | 20026202 | rs565344963 | G | A | 0.112151 | 0.010696 | 0.511987 |
| 11 | 15 | 20026205 | rs533002721 | A | T | 0.112476 | 0.0107821 | 0.507315 |
| 12 | 15 | 20026208 | rs12900467 | T | C | 0.112476 | 0.0107821 | 0.507315 |
| 13 | 15 | 20027647 | rs8029999 | C | G | 0.122335 | 0.00816835 | 0.817027 |
| 14 | 15 | 20028363 | rs74485484 | A | G | 0.117248 | 0.0102801 | 0.50935 |
| 15 | 15 | 20028762 | rs12903976 | C | T | 0.114862 | 0.00984186 | 0.535829 |
| 16 | 15 | 20029688 | rs34390178 | C | G | 0.134265 | 0.0296249 | 0.0493136 |
| 17 | 15 | 20030951 | rs35979813 | T | C | 0.116333 | 0.0104721 | 0.489063 |
| 18 | 15 | 20031739 | rs12442841 | T | G | 0.115869 | 0.0106059 | 0.485068 |
| 19 | 15 | 20034803 | rs144043507 | C | T | 0.13777 | 0.0270265 | 0.0665972 |
| 20 | 15 | 20034919 | rs78737532 | T | C | 0.115742 | 0.010556 | 0.487632 |
| 21 | 15 | 20039239 | rs12439778 | A | G | 0.116222 | 0.0099177 | 0.509795 |
| 22 | 15 | 20039935 | rs71464559 | T | A | 0.258699 | 0.0223527 | 0.0491104 |
| 23 | 15 | 20040322 | rs199772085 | A | G | 0.10038 | 0.0129446 | 0.442101 |
| 24 | 15 | 20040573 | rs12439263 | T | A | 0.117796 | 0.0093258 | 0.534502 |
| 25 | 15 | 20040668 | rs193121394 | G | A | 0.138654 | 0.0268971 | 0.065546 |
| 26 | 15 | 20042094 | rs12592301 | C | T | 0.256431 | 0.0219884 | 0.0644688 |
| 27 | 15 | 20044198 | rs28535042 | T | C | 0.259195 | 0.0222399 | 0.0619445 |
| 28 | 15 | 20044342 | rs117764863 | G | A | 0.0568624 | 0.00119022 | 0.955402 |
| 29 | 15 | 20044383 | rs35261146 | C | T | 0.116351 | 0.00980477 | 0.511018 |
| 30 | 15 | 20045025 | rs36028981 | G | A | 0.116646 | 0.00983351 | 0.510217 |
| ⋮ | ⋮ | ⋮ | ⋮ | ⋮ | ⋮ | ⋮ | ⋮ | ⋮ |
GeneticsMakie.mungesumstats!(gwas)
loci = GeneticsMakie.findgwasloci(gwas)
1 rows × 3 columns
| CHR | BP | P | |
|---|---|---|---|
| String | Int64 | Float64 | |
| 1 | 15 | 84419314 | 4.24936e-8 |
GCTA-COJO is planned to be implemented in the future for findgwasloci
GeneticsMakie.findgwasloci(gwas; p = 1e-6)
2 rows × 3 columns
| CHR | BP | P | |
|---|---|---|---|
| String | Int64 | Float64 | |
| 1 | 15 | 84419314 | 4.24936e-8 |
| 2 | 15 | 67467297 | 2.48762e-7 |
GeneticsMakie.findclosestgene(loci, gencode)
1 rows × 4 columns
| CHR | BP | gene | distance | |
|---|---|---|---|---|
| String | Int64 | String | Int64 | |
| 1 | 15 | 84419314 | TUBAP4 | 10363 |
GeneticsMakie.findclosestgene(loci, gencode; start = true)
1 rows × 4 columns
| CHR | BP | gene | distance | |
|---|---|---|---|---|
| String | Int64 | String | Int64 | |
| 1 | 15 | 84419314 | TUBAP4 | 10778 |
GeneticsMakie.findclosestgene(loci, gencode; proteincoding = true)
1 rows × 4 columns
| CHR | BP | gene | distance | |
|---|---|---|---|---|
| String | Int64 | String | Int64 | |
| 1 | 15 | 84419314 | ADAMTSL3 | 96474 |
loci = GeneticsMakie.findclosestgene(loci, gencode; start = true, proteincoding = true)
1 rows × 4 columns
| CHR | BP | gene | distance | |
|---|---|---|---|---|
| String | Int64 | String | Int64 | |
| 1 | 15 | 84419314 | ADAMTSL3 | 96474 |
function locuszoom(genes)
for gene in genes
@info "Working on $gene gene"
window = 1e6
chr, start, stop = GeneticsMakie.findgene(gene, gencode)
range1, range2 = start - window, stop + window
gwas_subset = gwas[findall((gwas.CHR .== chr) .& (gwas.BP .>= range1) .& (gwas.BP .<= range2)), :]
@info "Plotting LocusZoom"
f = Figure(resolution = (306, 792))
axs = [Axis(f[i, 1]) for i in 1:3]
GeneticsMakie.plotlocus!(axs[1], chr, range1, range2, gwas_subset)
GeneticsMakie.plotlocus!(axs[2], chr, range1, range2, gwas_subset; ld = kgp)
for i in 1:2
Label(f[i, 1, Top()], "Inguinal hernia (2022)", textsize = 6, halign = :left, padding = (7.5, 0, -5, 0))
rowsize!(f.layout, i, 30)
end
rs = GeneticsMakie.plotgenes!(axs[3], chr, range1, range2, gencode; height = 0.1)
rowsize!(f.layout, 3, rs)
GeneticsMakie.labelgenome(f[3, 1, Bottom()], chr, range1, range2)
Colorbar(f[1:2, 2], limits = (0, 1), ticks = 0:1:1, height = 20,
colormap = (:gray60, :red2), label = "LD", ticksize = 0, tickwidth = 0,
tickalign = 0, ticklabelsize = 6, flip_vertical_label = true,
labelsize = 6, width = 5, spinewidth = 0.5)
Label(f[1:2, 0], text = "-log[p]", textsize = 6, rotation = pi / 2)
for i in 1:3
vlines!(axs[i], start, color = (:gold, 0.5), linewidth = 0.5)
vlines!(axs[i], stop, color = (:gold, 0.5), linewidth = 0.5)
end
for i in 1:2
lines!(axs[i], [range1, range2], fill(-log(10, 5e-8), 2), color = (:purple, 0.5), linewidth = 0.5)
end
rowgap!(f.layout, 5)
colgap!(f.layout, 5)
resize_to_layout!(f)
save("./$(gene)-locuszoom.png", f, px_per_unit = 4)
end
end
@time locuszoom(loci.gene)
73.092486 seconds (245.55 M allocations: 12.107 GiB, 3.10% gc time, 83.83% compilation time)
┌ Info: Working on ADAMTSL3 gene └ @ Main In[16]:3 ┌ Info: Plotting LocusZoom └ @ Main In[16]:8
@time locuszoom(loci.gene)
10.318701 seconds (57.06 M allocations: 2.102 GiB, 3.77% gc time)
┌ Info: Working on ADAMTSL3 gene └ @ Main In[16]:3 ┌ Info: Plotting LocusZoom └ @ Main In[16]:8

Hypothetical scenario: you have run a GWAS and would like to visualize genome-wide significant loci automatically with 100s of GWAS results and functional genomic annotations
mungesumstats! function)findgwasloci function)Checkout example code!
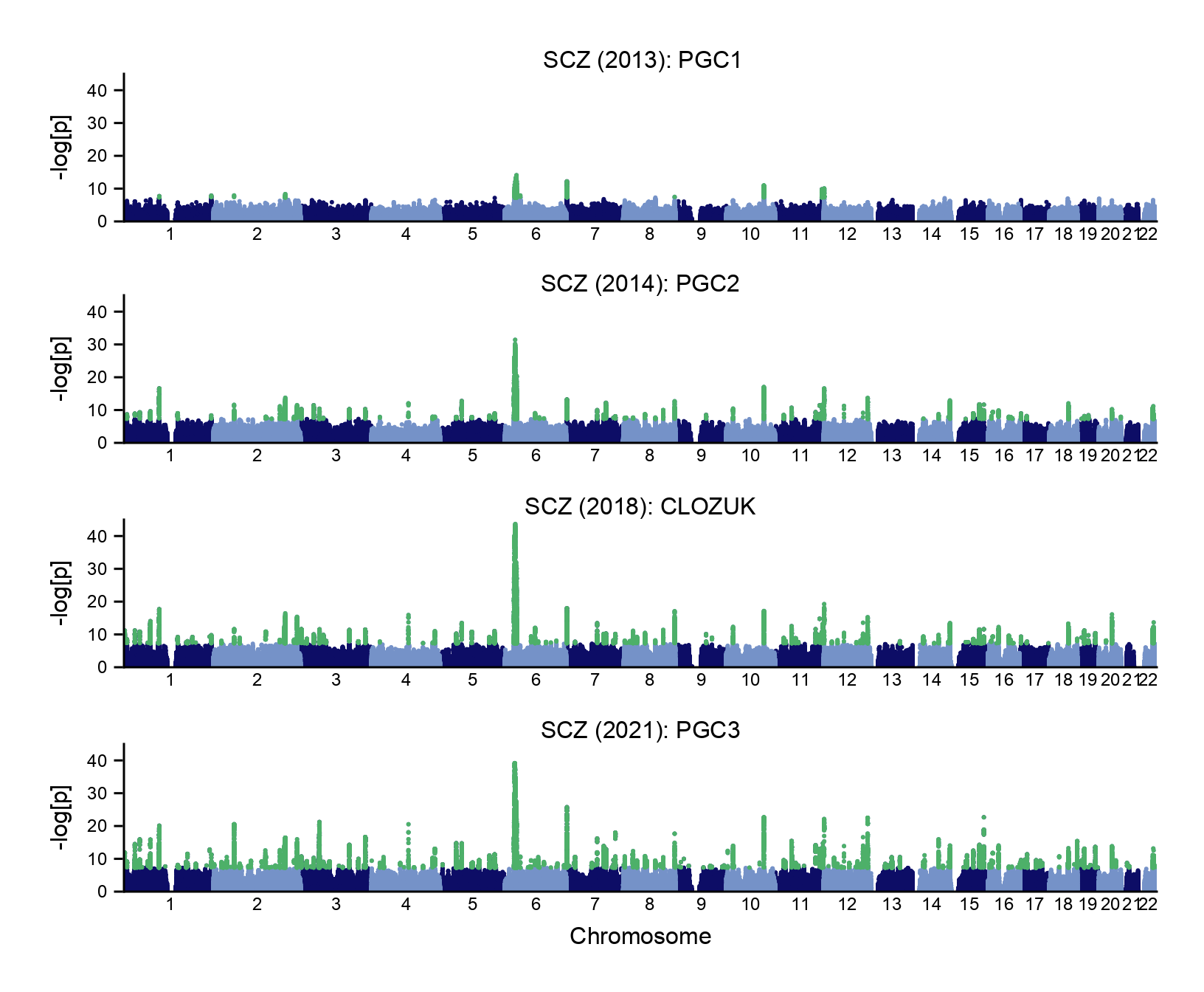
An increase in sample size has led to many more dicovery of GWAS loci for schizophrenia